Aktuální genetika
Multimediální učebnice lékařské biologie, genetiky a genomikyGenetika komplexních znaků
Genetická složka komplexních onemocnění
Příčinou současného prudkého nárůstu prevalence diabetu a dalších "civilizačních" onemocnění evidentně není markantní změna genetického fondu lidstva, ale spíš se jedná o důsledek měnící se složky prostředí, zejména pak nástupu tzv. západního životního stylu, tj. především zvýšeného energetického příjmu a snížené míry fyzické aktivity. Nicméně tento stav vyplývá z kombinovaného (interaktivního) působení změny prostředí, která se projevuje působením na citlivý genotyp. Na přítomnost genetické složky v patogenezi diabetu ukazuje několik pozorování:
1. Hromadění (agregace) znaku v rodinách
Pokud má nějaký znak zřetelnou tendenci k častějšímu výskytu v rodinách, jedním z možných důvodů je vyšší míra sdílení genů mezi členy rodiny, z nichž některé predisponují svého nositele k danému onemocnění. Je zřejmé, že samotný fakt častějšího výskytu znaku v rodinách není důkazem pro existenci významní genetické složky. Na tu ukazují mj. i další indicie. Jednou z nich může být odhad rizika manifestace znaku u příbuzných diabetiků s koeficientem příbuznosti 0,5 (děti, sourozenci). Například má-li jeden rodič diabetes 2. typu, je přibližně 40% pravděpodobnost, že se diabetes vyvine u jeho potomka. Jsou-li postiženi oba rodiče, riziko se zvyšuje na 70%. Navíc se ukazuje, že i nediabetičtí příbuzní diabetiků mají zhoršenou inzulínovou senzitivitu a vyšší hladinu inzulínu oproti kontrolám.
2. Studie dvojčat
Dvojčata jsou jedinečným "experimentem přírody na lidech" a studium souborů dvojčat přispělo značnou měrou k prvotní identifikaci genetické složky řady onemocnění. Jednovaječná (monozygotická - MZ) dvojčata vznikají rozštěpením jedné zygoty a jsou tedy geneticky identická (s výjimkou náhodné inaktivace X-chromozómu u dvojčat ženského pohlaví) a sdílejí rovněž i intrauterinní prostředí. Naproti tomu dvojvaječná (dizygotická - DZ) dvojčata vzešlá z oplodnění dvou různých oocytů sdílejí navzájem jen jednu polovinu genů (stejně jako jakýkoli sourozenecký pár). U studií dvojčat se zjišťuje konkordance, tedy shoda a diskordance - neshoda uvnitř páru dvojčat pro sledovaný znak, tedy např. diabetes. Porovnáním konkordance MZ a DZ dvojčat je možné odhadovat poměr genetické a negenetické determinace daného znaku, kde platí, že pokud nalezneme větší konkordanci mezi MZ dvojčaty ve srovnání s DZ dvojčaty, svědčí to pro přítomnost genetické složky sledovaného znaku. Pokud konkordance mezi MZ dvojčaty naopak není úplná, považuje se to za indikaci úlohy negenetických faktorů. Zajímavým zjištěním jedné studie bylo, že když byla opakovaně vyšetřena skupina MZ dvojčat patnáct let po původní studii, 76% párů původně klasifikovaných jako diskordantní pro daný znak se stalo konkordantními.
3. Etnická variabilita
Při globálním pohledu je možné zjistit mnohonásobně rozdílnou frekvenci mnohých komplexních onemocnění mezi jednotlivými etniky. Některé etnické skupiny vykazují extrémně vysokou prevalenci civilizačních chorob, která je vysvětlována recentní selekcí tzv. "úsporného genotypu" mechanismem přežívání jedinců nejodolnějších vůči často se střídajícím obdobím nedostatku a nadbytku potravy. Námitce, že část rozdílu frekvence civilizačních onemocnění mezi jednotlivými etniky padá na vrub negenetickým, kulturně-sociálním faktorům, protiřečí fakt, že rozdíly můžeme pozorovat i mezi skupinami s podobným životním stylem a dietními návyky.
Metody a úskalí zkoumání genetické komponenty komplexních onemocnění
V zásadě jsou používány dva postupy identifikace genů zodpovědných za komplexní onemocnění. První způsob vychází z již existující znalosti vztahu daného genu/proteinu ke konkrétnímu onemocnění anebo se takový vztah dá vzhledem k dosud získaným poznatkům o jeho funkci či struktuře předpokládat. Takový "funkční" kandidát je pak podroben detailní genetické a molekulárně biologické analýze a jsou testovány hypotézy o asociaci / kosegregaci specifických variant (polymorfismů) daného genu se zkoumaným znakem, tedy přímo diabetem nebo některým z tzv. přechodných fenotypů. Tento postup je v literatuře nazýván "functional cloning" (v češtině odpovídá nepříliš používanému výrazu funkční klonování). Oproti tomu druhá metoda vychází výlučně z identifikace oblasti genomu, kde se na základě vazebných studií nachází místo (lokus), který významně ovlivňuje variabilitu zkoumaného znaku. V místě nejsilnější vazby se pak v ideálním případě molekulárně genetickými metodami shrnutými v pojmu poziční klonování ("positional cloning") podaří identifikovat zodpovědný gen, resp. jeho alelu kauzálně spjatou se segregujícím znakem. Díky prudkému nárůstu počtu známých genů, ke kterému došlo v důsledku rozsáhlých projektů sekvenace genomů člověka a modelových organismů, se obě metody nejčastěji používají zároveň, když jsou identifikovány pravděpodobné kandidátní geny v úseku genomu, jehož spojitost se zkoumaným znakem ukázala vazebná analýza. Pro tuto strategii se používá "pozičně-kandidátní přístup" (positional candidate gene approach). Teoretické aspekty genetické analýzy komplexních znaků jsou detailně popsány v dostupných publikacích (Hatina a Sykes, Lékařská genetika 1999), takže se omezíme jen na základní charakteristiku dvou majoritních postupů:
1) Vazebná analýza
Pro vazebnou analýzu (linkage mapping) komplexních znaků jsou nejčastěji užívané tzv. neparametrické metody, tedy takové, kde a priori nepředpokládáme žádný konkrétní způsob dědičnosti, počet genů nebo míru vlivu negenetických faktorů. U vybrané skupiny rodin s frekventním výskytem onemocnění, případně u souboru sourozenců sdílejících určitý fenotyp zkoumáme, jestli postižení jedinci významně častěji nesdílejí některé alely. Toto testování předpokládá znalost genotypu pro stovky míst v genomu u jedinců ze souboru. Pro genotypizaci se nejčastěji používají stovky anonymních markerů rozmístěných po celém genomu, charakterizujících polymorfismy typu mikrosatelitů (krátkých repetic di-, tri- nebo tetranukleotidů) s vysokou mírou heterozygocie, která je základem pro informativnost analýzy.
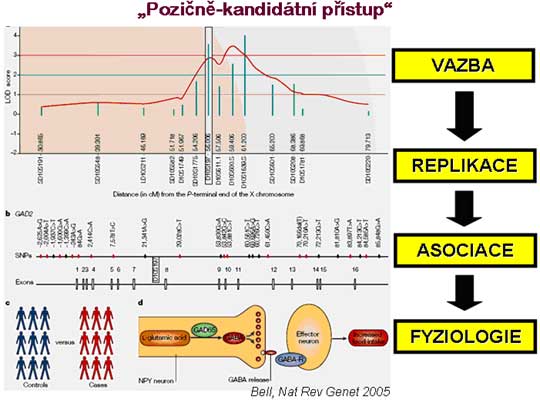
2) Asociační studie
Asociační studie se přímo zaměřují na testování vztahu mezi konkrétní alelou, genotypem nebo haplotypem (souborem úzce vázaných genotypů) a onemocněním. Většinou mají asociační studie charakter "case-control" (případ-kontrola), kdy je porovnáváno relativní zastoupení určitého polymorfismu mezi skupinami, z nichž jedna dané onemocnění vykazuje (diabetici), zatímco druhá nikoli. Pokud je i při zachování zásad správného výběru obou skupin a po statistickém odfiltrování falešně pozitivních výsledků nalezena asociace mezi zkoumanou genetickou variantou a onemocněním, lze vyslovit předpoklad, že tato alela nějakým způsobem s onemocněním souvisí, nebo je ve vazebné nerovnováze (linkage disequilibrium) se skutečnou kauzální alelou a slouží tedy víceméně jako její marker. Je samozřejmě možné takto testovat polymorfismy čistě funkčních kandidátních genů nebo takových, na které ukázala předchozí vazebná analýza. Z principu této strategie jsou zřejmá i její omezení a tak je obecně nutné konkrétní výsledek replikovat alespoň v jedné nezávislé studii, aby byl považován za biologicky významný. Určitou renesanci asociační analýza prožívá s nástupem nových technologií, kdy např. při použití čipů umožňujících současné testování desítek až stovek tisíc jednonukleotidovýcch polymorfismů (SNP, viz níže) se stává realizovatelnou vpravdě celogenomová asociační analýza, i když s množstvím testovaných polymorfismů prudce narůstá riziko falešně pozitivních pozorování a je třeba velké opatrnosti při interpretaci výsledků těchto studií.
Úskalí genetiky komplexních znaků
Zásadním problémem při zkoumání genetické složky multifaktoriálních onemocnění je samotné vymezení analyzované nosologické jednotky. Mnoho "diagnóz" (autismus, obezita, hypertenze apod.) představuje souhrn geneticky odlišných onemocnění. Mezi hlavní problémy analýzy i proto patří:
1) Oligogenní až polygenní dědičnost komplexních onemocnění.
Metody genetické analýzy doposud aplikované na komplexní onemocnění byly povětšinou vytvořeny pro mapování monogenních znaků, jsou tedy založeny na matematických modelech předpokládajících existenci jediného lokusu s výrazným efektem na variabilitu zkoumaného znaku. U komplexních onemocnění se naopak předpokládá, že jejich genetická komponenta je výslednicí působení více genů, které interagují mezi sebou a s faktory vnějšího prostředí (oligogenní epistatický model), každý z nich však variabilitu znaku ovlivňuje pouze málo.
2) Genetická heterogenita
Může být dvojího druhu. První z nich je heterogenita samotného znaku, kdy jedna "diagnóza" zahrnuje několik geneticky oddělených entit, jak bylo zmíněno výše. Tomu se částečně dá čelit přechodem od kvalitativní analýzy (tedy dichotomického dělení nemocný/zdravý) k analýze kvantitativní, využívající některých z přechodných fenotypů úzce vázaných k samotnému onemocnění, např. specifických biochemických vyšetření. Druhou formou je potom tzv. alelická heterogenita, tedy asociace mezi daným znakem (onemocněním) a dvěma či více alelami stejného lokusu. Tyto faktory musí být samozřejmě reflektovány v použití nových statistických modelů a postupů, nikoli matematického aparátu, který kalkuluje s jednoduchým monogenním typem dědičnosti. Jsou proto vyvíjeny různé varianty shlukovacích, faktorových či Bayesiánských analýz.
3) Různý věk nástupu (age of onset),
díky němuž je možné, že u jedinců s nediagnostikovaným onemocněním v době provádění studie dojde v průběhu času k manifestaci geneticky determinovaného znaku, což snižuje informativnost skupiny takto klasifikovaných jedinců.
4) Neúplná penetrance
Stav, kdy ne u všech nositelů disponujících alel dojde k manifestaci daného onemocnění, ať už proto, že je genetická dispozice "titrována" ochranně působícími alelami dalších genů a interakcí, případně příznivým vlivem složky prostředí (např. životní styl).
5) Etnická heterogenita
Dosavadní výsledky analýz genetické komponenty multifaktoriálních onemocnění potvrzují předpoklad, že pro populace různých etnik se budou skupiny odpovědných genů do určité míry lišit a význam jednotlivých variant bude mít v různých populacích jinou míru relevance pro výsledný fenotyp - pro ošetření etnické heterogenity kohorty je pak nutná buď její stratifikace nebo použití speciálních statistických modelů zahrnujících předpoklad tzv. genetického "přimísení" (admixture). Podstatně menší heterogenitu vykazují tzv. populační izoláty, tedy populace izolované geograficky, kulturně nebo nábožensky, které v mnohých případech historicky vznikly jen z velmi omezeného počtu původních zakladatelů (viz např. Hamet et al., 2005).
6) Fenokopie
Projev daného fenotypu u jedinců, kteří nezdědili příslušnou sadu patologických alel - např.v důsledku .
Komplexní znaky u eperimentálních modelů
Klasická strategie pro identifikaci genetických determinant komplexních onemocnění u experimentálních modelů by se dala shrnout asi takto: vycházíme ze dvou inbredních kmenů (potkana, myši), tedy takových kmenů, kde všichni jedinci (téhož pohlaví) každého z kmenů jsou geneticky identičtí. Tyto dva parentální kmeny se liší ve zkoumaném znaku - např. hypertenzní kmen vs. normotenzní kmen. Pokud jedince těchto dvou kmenů navzájem křížíme, jejich potomstvo (první filiální generace - F1) je heterozygotní v rozsahu celého genomu. Dále máme dvě možnosti, buďto postupujeme cestou zpětného křížení (tedy křížení F1 s parentální generací) nebo křížíme navzájem dvojice F1 a získáme populaci druhé filiální generace (F2, zpravidla několik set jedinců). V obou případech dosáhneme stavu, kdy v experimentální skupině segregují jak alely obou parentálních kmenů, tak i vlohy pro zkoumaný znak (v našem příkladě hypertenzi). V dalším kroku je třeba, abychom celou populaci zpětných kříženců (backcross) nebo F2 kříženců (intercross) charakterizovali z hlediska fenotypu (např. stanovení denního profilu systolickího a diastolického krevního tlaku), tak z hlediska genotypu. Nejčastěji se používá sada několika set tzv. mikrosatelitních markerů, které jsou rovnoměrně rozmístěny po celém genomu. U každého jedince F2 zjistíme, jestli zdědil obě alely daného markeru od jednoho či druhého progenitora (parentálního kmene), příp. jestli má po jedné alele od každého a je tedy pro daný úsek genomu heterozygotní (u zpětného křížení samozřejmě připadá v úvahu jen jeden typ homozygota nebo heterozygotní stav). Když máme k dispozici tyto charakteristiky, zkoumáme (za pomoci specializovaného software), jestli některé alely nejsou výrazně předávány potomkům současně s dispozicí pro vysoké nebo naopak nízké hodnoty sledovaného kvantitativního znaku.
Nejjednodušší variantou je porovnání hodnot např. inzulinémie mezi skupinami rozdělenými podle genotypu postupně v každém markeru. Pokud budou jednotlivé genotypové třídy vykazovat signifikantně rozdílné parametry krevního tlaku, lze předpokládat, že daný marker, případně variace DNA v jeho blízkosti nějakým způsobem krevní tlak ovlivňuje. Tato metoda, nazývaná "marker regression", má ale několik nevýhod. Předně, při testování mnoha set (a dnes mnohdy několika tisíc) markerů je pravděpodobné, že alespoň u jednoho či několika náhodně odlišnou distribuci kvantitativního znaku objevíme. Abychom se vyhnuli takovým falešně pozitivním nálezům, je potřeba patřičně upravit hladinu signifikance. Další zásadní nevýhodou tohoto přístupu je, že testujeme asociaci jen přímo v místech, ve kterých se nacházejí námi typizované markery, což v experimentu běžných parametrů znamená pomíjení desítek megabází DNA mezi nimi. Proto se nejčastěji používá metoda tzv. intervalového neboli QTL (quantitative trait locus) mapování, která umožňuje odhadovat genotyp i v úsecích mezi námi genotypizovanými markery. Pokud se QTL (tedy lokus, který výrazně ovlivňuje variabilitu ve zkoumaném znaku) podaří identifikovat, nastává fáze pozičního klonování, která je v principu velmi podobná strategii popsané u studií lidských.
Obezita, definovaná jako body mass index (podíl hmotnosti a druhé mocniny tělesné výšky) větší než 30 kg/m2, je typickým příkladem komplexního onemocnění. Je nasnadě, že se na vzniku obezity podílí značnou měrou složka prostředí, především nerovnováha mezi energetickým příjmem a výdejem, ale ukazuje se, že nemalou úlohu hrají i faktory genetické.
Mapa doposud zjištěných genetických variant asociovaných s obezitou jak u člověka, tak u modelových organismů, je k dispozici na internetu na níže uvedené adrese:
