Current genetics
Multimedia E-textbook of medical biology, genetics and genomicsIntroduction to Proteomics
Proteomics if a field of global study of the expression of genetic information at the protein level (proteome). It also deals with assessment of three-dimensional structure of proteins and their interactions.
Human Proteome Organization (HUPO, www.hupo.org) has postulated in 2001 the aims of proteomics in a more precise manner as identification of all proteins coded by human genome (and other genomes, especially those of model organisms) with the subsequent assessment of a) their (protein) expression in different cell types of the organism - expression proteomics, b) protein distribution in subcellular compartments of the organelles, c) post-translational modifications of the proteins, d) protein-protein interactions (b-d constitute the structural proteomics) and e) relation between protein structure and function (functional proteomics).

The wide range of the definition of proteomics precipitates into an array of distinct sub-specialties, e.g. Clinical proteomics is trying to identify via the analysis of the protein spectrum in the biological samples (urine, saliva, blood) new markers that can be used in future early diagnosis and treatment of a specific disease. The major role in all spheres in early diagnosis and treatment of a given disease is played by bioinformatics (digitalization, 2D picture analysis of the gels, modeling 3D protein structures and the networks of protein interactions, creation, care and development of publicly available databases). One of the interesting proteomic projects is Swedish Human Protein Atlas (http://www.proteinatlas.org/), that groups the information about expression of individual proteins in different tissues but its uniqueness lies in the accompanying archive of immunohistochemically stained samples - at the time of writing of this text, there were more than 400,000 available. Even though it may seem that because of the availability of RNA/cDNA microarray chips for analysis of expression on the level of transcripts there is enough detailed information about the currently ongoing status of expression of genetic information, it is necessary to understand that
- not all mRNAs will be translated into the protein
- The level of transcription of specific, protein-coding RNA not always corresponds to the level of expression and moreover, to activity of the coded protein due to many factors (mRNA, RNA splicing, posttranslational protein modifications, etc.)
Similar to other "-omics" the development of proteomics was significantly influenced by the recent developments in technology. The real development of the proteomics started only after the classical method of 2D electrophoresis was joined by "soft" ionization techniques of mass spectrometry. These techniques are performed using matrix assisted laser desorption/ionization (MALDI) and electrospray ionization (ESI).
Figure 1: Major proteomics directions
Proteomic methods
1. 2D protein electrophoresis
Nowadays the two-dimensional (2D) protein electrophoresis is a method of choice for protein separation. This method enables to distinguish up to 10 000 proteins. The method is based on two distinct physical and chemical features of proteins: first, the proteins are separated according to their isoelectric point (pI). Isoelectric point is such a pH value, where the overall protein charge equals to zero. This can be obtained by creating a pH gradient in the gel where protein is loaded and electric current is applied. Proteins then migrate towards cathode or anode according to their total charge up to the point where the gel pH equals pI of a given protein. After the separation in the first direction, common electrophoresis on a polyacrylamide gel (PAGE) is applied, but the electric current is applied perpendicular to the original orientation of electrodes. Proteins then migrate in the second direction through gel only according to their size. After both phases of 2D electrophoresis, it is necessary to visualize proteins by one of the staining or labeling methods (chemical or radioactive). The resulting "maps" of proteins can be compared for example in between the experimental and control sample or among the samples from patients with specific disease and their healthy controls and thus identify differentially expressed proteins that can be linked with the pathogenesis of the studied disease. The identity of differentially expressed proteins is verified after "cutting out" the area of the gel with given difference and subsequent analysis using mass spectrometry.
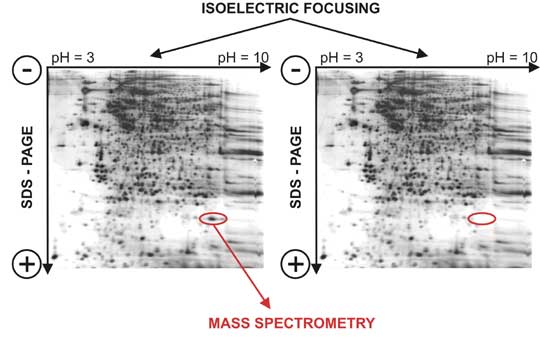
Figure 2. 2D electrophoresis. After isoelectric focusing in pH gradient of the electric field the electrophoretic separation is used in polyacrylamide gel (SDS-PAGE….sodium dodecylsulphate - polyacrylamide gel electrophoresis). Proteins can be visualized after staining (on the picture in Coomasie blue).
2. Mass spectrometry (MS)
Mass spectrometry is a method that enables precise measurement of molecular weight of a broad spectrum of substances. As the studied substance has to be intact in gas phase, the use of mass spectrometry for protein analysis (but also of polysaccharides and oligonucleotides) was only enabled by development of "soft" ionization techniques of mass spectrometry, like matrix assisted laser detection of desorption/ionization (MALDI) and electrospray ionization (ESI). Protein identification is generally performed in two ways:
- Protein is digested by trypsin or by other proteolytic enzyme to smaller peptides and their precise molecular weights are measured using MS. The spectrum of those molecular weights is then compared with theoretical spectra that are calculated from protein sequences from available databases (using bioinformatics tools).
- Tandem MS enables to choose the peptide which is then fragmented by the collision with inert gas. The fragmentation pattern gives either full of partial information about protein sequence that is subjected to the search in databases.
Apart of protein identification, MS is a priceless tool for protein posttranslational modification analysis, because it enables to localize given modifications within the protein and also helps to find out the nature of such modification.
Proteomics in medicine
As stated above, one of the main goals of proteomics is the identification of novel markers that can be used for prediction, prevention, diagnosis, prognosis and therapy optimalization in human disease. To use proteomics in real everyday clinical practice it is necessary that the biological material where such marker would be analyzed is easily accessible (blood, urine, salive, cereblospinal fluid). The other way to use proteomics is mainly within the experiment, one of the emerging applications is in the field of therapeutical drug discovery and pharmacoproteomics.
Sources/Other literature
- Molecular Biologist's Guide to Proteomics
Molecular Biologist's Guide to Proteomics