Current genetics
Multimedia E-textbook of medical biology, genetics and genomicsNon-Mendelian Heredity
Some diseases and traits coded by specific gene variants do not follow the laws of penetrance into next generations as "classical" monogenic diseases with autosomal or gonosomal inheritance. This heterogeneous group is specified by non-Mendelian inheritance. Into this group fall the mitochondrial inheritance, instability of repetitive sequences and genomic imprinting. Totally separate stands the epigenetics (see chapter Epigenetics). The penetrance and expressivity of the trait that complicates the monogenic inheritance do not fall into this group.
Mitochondria and mitochondrial inheritance
a) Mitochondrial DNA
Even though majority of the human genome is stored within the nuclear DNA, 37 structural genes are coded in the circular mitochondrial genome, that has 16569 base pairs of DNA (mtDNA). One third of those genes code subunits of respiratory chain complex responsible for ATP production within the oxidation-phosphorylation system (OXPHOS, 13 polypeptides, the rest is coded by nuclear genome). Other mitochondrial genes code specific RNA molecules necessary for protein synthesis (22 transfer RNAs and 2 ribosomal RNAs). There are several hundreds or thousand copies of mtDNA in cytoplasm and in every mitochondrion there are several mtDNA molecules. The homoplasmy is when there is just one homogeneous mtDNA population (this is true for majority of "normal" individuals). The mitochondrial frequency of mutations is 10-20x higher compared to nuclear DNA, which leads to heteroplasmy, where there is a mixed population of more variants of mtDNA. Many features of mtDNA support the theory about the origin of mitochondria as endosymbiotic bacteria of proto-eukaryotic cells more than 1.5 billion years ago. The most remarkable features are circular genome organization, the absence of histones, and the absence of introns or discrete beginnings of replication. Moreover mitochondria of vertebrates and other organisms possess the variations from the universal genetic code - UGA codon is not a termination codon, but codes for tryptophan, on the other hand codons AGA and AGG do not code for leucine, but terminate translation and the codon AUA codes for methionine instead for leucine. Biologically closest relatives are Rickettsias (the cause of typhus) that are obligatory intracellular parasites. It is presumed that Rickettsias and mitochondria have a common ancestor that made the transition from autonomous existence to endosymbiosis.
Mitochondrial DNA really contains one important "non-coding" sequence, so-called D-loop (a short part of mtDNA, where the heavy chain is pushed out by DNA fragment (500-700 nucleotides), complementary to the light chain (in this region mitochondria has triple-stranded DNA). Here is the beginning of replication of so called heavy chain, the fragment works as a primer for the beginning of replication. The beginning of replication of light chain is placed out of the D-loop, approximately in 2/3 of mtDNA. Transcripts of both chains have to be cleaved to release the functional RNA (rRNA, tRNA and mRNA).
Probably the the best source of information about mitochondrial genome, mitochondrial diseases and other related themes can be found on MITOMAP:A human mitochondrial genome database, http://www.mitomap.org/.
b) Mitochondrial inheritance
From the genetics point of view the crucial fact is, that mtDNA comes to the next generation only from mother (matrocline inheritance) as after conception only mitochondria of the egg are preserved. This is not only probably due to the imbalance in the number of mitochondria of human oocyte (app. 100 000) and sperm (50-70), it is probably an active process, that after conception eliminates mitochondria of paternal origin. Due to this fact we can observe a typical maternal mode of inheritance of diseases caused by mutations of mtDNA in the pedigree (Fig.1). If heteroplasmic mutation is inherited or happens during the early phases of embryogenesis, both normal and mutated variant are randomly given to daughter cells during the cell division (mitotic and meiotic segregation). The distribution and percentage of mutated mtDNA is therefore dependent on the time of occurrence of the mutation and on the type of affected cell in different tissues.
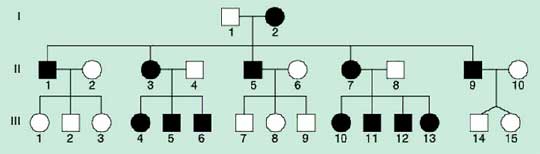
Fig.1 Typical pedigree for the disease with mitochondrial type of inheritance
c) Diseases with mitochondrial type of inheritance
It is necessary to bear in mind that not all disease caused by dysfunctional mitochondria have mitochondrial type of inheritance. If there is a defective protein and parts of its subunits are coded by genes of the nuclear genome and some by mtDNA, the mode of inheritance can be completely Mendelian. In their monography, Sršeň and Sršňová defined four basic characteristics of heritable diseases where mitochondrial inheritance should be presumed:
- Maternal mode of inheritance (see above)
- Thanks to mitotic and meiotic segregation we can find different level of affectedness in different tissue and variable phenotypic manifestations in offspring of one maternal line, which can be attributed to threshold effect (it is necessary for manifestation of dysfunction to overcome a certain ratio of mutant vs. normal mtDNA - in majority of cases 60-90%).
- Manifestations of OXPHOS system dysfunction. Hereditary mitochondrial diseases usually affect tissues with high needs for energy income as neuromuscular or central nervous system. Common symptoms of mitochondrial diseases are encephalopathy, ataxia, spasms, (cardio)myopathy, deafness or diabetes mellitus.
- The difference from X linked gonosomal dominant heritability is in the absence of transfer of mitochondrially linked disease to the offspring and in majority of affected women in X linked dominant inheritance.
Repetitive sequence instability
Repeat instability is a recently discovered type of mutation that causes more than 40 neurodegenerative and neuromuscular diseases. These mutations are sometimes called as dynamic as their number of repeats is changing from generation to generation and also from tissue to tissue. On the other hand "static" mutations are inherited practically in unchanged way. The most common form of repeat instability are the trinucleotide repetitions, but also diseases associated with tetranucleotide repetitions (dystrophia myotonica 2), pentanucleotide repetitions (spinocerebellar ataxia 10), and also repetitions of minisatellite and megasatellite sequences. In majority of diseases of this group more repeats mean sooner manifestation and more severe progression of the disease. The other characteristic feature of repeat instability is so called genetic (generation) anticipation, i.e. the possibility of growing number of repeats with growing number of generations. The genetic anticipation is usually specific for each disease for offspring of one gender. This way the children of healthy parents with elevated number of repeats but still below the "threshold" (the parents are carriers of so called premutation) can have a manifested disease.
Fragile X syndrome was the first described disease of this group. The Fragile X syndrome is the second most common cause of inborn mental retardation (the most common is Down syndrome). Phenotype of this disease is very variable; apart from mental deficit of different degree the affected individuals can express these features: behavior problems resembling autism, high voice, prolapsed mitral valve, macrocephaly, specifically long face with prominent mandible, big ears, macroorchidism in affected males. Even though this syndrome was first described in 1943 (Martin et Bell), it was named by the observation of Herbert Lubs, that in 1969 described secondary constrictions of long arm of chromosome X in several mentally retarded men, the fragile region (Xq27.3). The molecular cause of majority cases of Fragile X syndrome is the trinucleotide expansion CGG in 5'-UTR of FMR1 gene (Fragile X Mental Retardation 1) that leads to silenced transcription and missing product of FMR1 gene - the FMRP protein. The full mutation is when the number of repeats is higher than 200, the normal number of repeats is 6-50. The premutation is here described as 60-200 repeats, only recently it was discovered that this variant is responsible for syndrome called Fragile X tremor/ataxia syndrome (FXTAS). The absence of FMRP protein is linked with a huge number of pathophysiological processes mainly in the nervous system, e.g. the presence of many long "immature" dendrites and defective signal transduction on postsynaptic level in metabotropic glutamate receptors.
Friedreich ataxia (FA) is the most common form of hereditary ataxia. Majority of patients have homozygous expansion (GAA)n in the first intron of frataxin gene that causes lower mRNA and protein levels. More trinucleotide repeats are present, less mRNA and protein is produced. Lower concentrations of frataxin are followed by iron accumulation in mitochondria that causes higher sensitivity to oxidative stress and lowering of oxidative phophorylation.
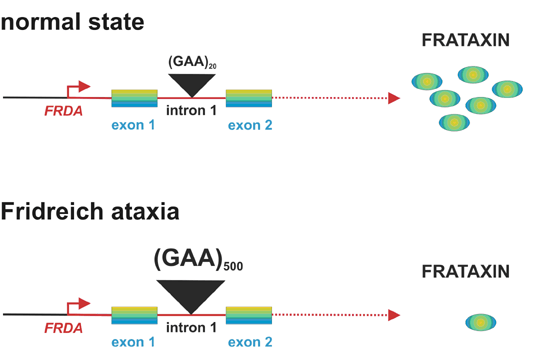
Fig. 2 Modified from Gatchel and Zhogbi, 2005.
Further sources:
- Genetics of developmental disabilities. Butler MG., Meaney F.J. eds.. Taylor & Francis Group, LLC. 2005
- MITOMAP: A Human Mitochondrial Genome Database. http://www.mitomap.org, 2005.