Current genetics
Multimedia E-textbook of medical biology, genetics and genomicsGender & Genetics
Sex determination
In humans, the sex is strictly genetically determined. This became evident through studies of Turner and Klinefelter syndromes. Individuals with Turner syndrome, a cytogenetic abnormality with karyotype 45, X, develop as women: have all Müller duct derivatives (uterus and oviducts), do not have Wolffian duct derivatives, have ovaries and develop as women psychosocially. Nevertheless, they have a lot of pathological features. Much of the pathology can arise from failure of oocyte meiosis in pachytene (during the fetal development) probably due to errors in handling the unpaired X chromosome. The oocytes are progressively lost and the ovaries do no contain follicles, which means no hormone production, no endometrium cycling, impaired pubertal development, premature atherosclerosis and osteoporosis. This means that the lack of Y chromosome leads to female sex development. The impairment of pubertal sexual development is secondary due to oocyte depletion.
Individuals with Klinefelter syndrome have karyotype 47, XXY. Despite two X chromosomes, of which one becomes inactivated (as in females with normal karyotype) the individuals with such karyotype are males, although they have problems with fertility and androgen deficiency.
From these two syndromes, it can be concluded that Y is the male determining chromosome - whenever this chromosome is present the male developmental program is initiated, when the chromosome is lacking, a female developmental pathways are initiated. Moreover, the dosage of the X chromosome does not influence sex determination.
Through analysis of human and mouse sex reversal syndromes (XX males and XY females) it was shown that a single gene Sry (Sex determining region on chromosome Y) is responsible for the initiation of male development. Sry gene is located in the male specific portion of the short arm of Y chromosome (figure Y chromosome), just downstream of pseudoautosomal region 1 (PAR1). Rarely, a crossing over event may occur not in the PAR1, as obligate, but in the neighboring sequence, which has some similarity to X chromosome. In that case, Sry gene is translocated to X chromosome and the result is 46, XX male.
Sry protein is a transcription factor of HMG (high mobility group) family. It is expressed in the supporting cells of the indifferent gonad and triggers their differentiation into Sertoli cells. Sertoli cells then organize into cords that encase the primordial germ cells (PGC) and induce their differentiation into prospermatogonia. These epithelial cords later form lumen and become seminiferous tubules.
Sry gene is not expressed only in testis, but also in the brain, in substantia nigra, where it upregulates expression of tyrosine hydroxylase, an enzyme necessary for dopamine synthesis. When the expression of Sry in substantia nigra is turned off in mice, the mice develop symptoms of Parkinson disease. The requirement of Sry for dopamin synthesis in men could explain their greater susceptibility to for development of Parkinson disease (relative risk is 1,5). In females, nubstantia nigra is probably smaller (proven in mice - there is 20% difference), but dopamin synthesis is controled by other, presumably more stable mechanisms.
Direct downstream target of Sry is Sox9 (Sex determining region Y [Sry]-related box-9), a transcription factor also belonging to HMG family, located on chromosome 17q24. Mutations in this gene cause campomelic dysplasia, a syndrome, in which XY female sex reversal (karyotype is 46, XY, but sex is female) is combined with skeletal dysplasia (Sox9 is also necessary for cartilage development). Duplication of Sox9 gene causes XX male sex reversal (karyotype 46, XX, male sex). Everything fits together - decreasing Sox9 function leads to female development, increasing Sox9 expression boosts the male pathway.
One recently identified pathway downstream of Sry is accumulation of glycogen in pre-Sertoli cells via insulin/insulin-like growth factor (IGF) stimulated glucose uptake. This energy storage is necessary for massive Sertoli cell proliferation and thus testis development in mammals. XY mice with mutations for all three insulin receptor members (Ir, Igf1r and Irr) developed ovaries and showed a completely female phenotype.
Shortly after induction by Sry, the Sertoli cells perform many tasks: First they induce differentiation of primordial germ cells (PGC) into sprematogonia - a process accompanied with formation of testis cords from Sertoli cells, which enclose germ cells. What is the nature of the differentiation signal transmitted to PGC (and the role of PGC themselves) is not yet clear enough, but prostaglandin D2 is one of the candidates. Second, the Sertoli cells also produce AMH (anti-müllerian hormone) which promotes disintegration of Müller duct, blocking thus development of Müller duct derivatives - uterus and oviducts. Third, Sertoli cells are responsible for Leydig cell differentiation. Leydig cells in turn produce androgens, which partially govern testis descent and are fully responsible for masculinization of external genitalia. After this fetal period of androgen production, the testosterone level decreases. The androgen level goes up again during pubertal development in males - which is also heavily androgen-dependent. Mutations in androgen receptor (AR) gene (on X chromosome) cause androgen insensitivity syndrome. If the function of androgen receptor is completely abolished, in the complete androgen insensitivity syndrome also dubbed "testicular feminization", the XY individuals develop female external genitalia, pubertal and psychosocial development is female too. However, as this is a post-testicular block, these females have testes, which produce androgens and descend out of the abdominal cavity (can be frequently found in the inguinal canal or in major labia). As the testes produce AMH, they do not have Müller duct derivatives - uterus and oviducts, so present as primary amenorhea. A special feature is missing pubic and axillary hair, as these are also induced by androgens.
Female sex differentiation is initiated in the primordial gonad when Sry is not expressed, as a default developmental pathway of the gonad. The primordial germ cells become oocytes precursors, proliferate, recruit follicular cells and undergo first meiotic division, but they stop in diplotene stage until ovulation. This especially long diplotene is called dictyotene. However, there is some new evidence, that oocytes can develop from germ stem cells de novo differentiated from coelomic epithelium of ovaries. Without this still controversional mechanism of oocyte renewal all oocytes go through the meiotic prophase in fetal ovaries. When, e.g. after 45 years, one follicle is selected and grows, the oocyte completes first meiotic division just before ovulation. After ovulation, second meiosis proceeds spontaneously up to metaphase II. There the division stops again and can go on only after fertilization.
Descent of the testis
Cryptorchidism is the most common disorder of sexual development in the newborn (about 3%, fortunately, substantial proportion recovers spontaneously - at 1 year it remains in less than 1% boys). Complications of impaired testicular descent include not only infertility but also increased risk for testicular malignant tumors.
Mutations of the mouse genes Insl3, Great (G-protein coupled receptor that affects testicular descent) and Hoxa10 result in male infertility secondary to cryptorchidism. The sexual dimorphic position of of the gonads is dependent on the dimorphic development of the two parts of genital mesentery, the cranial suspensory ligament (CSL) and gubernaculum (caudal genital ligament). In males, transabdominal phase of testicular descent is characterized by movement of the testis into the inguinal region, as a result of gubernaculum development and CSL regression. On the other hand, in females, CSL and not gubernaculum develops, keeping thus the ovary adjacent to kidney. During the second, inguinoscrotal phase, descent of the testis to the scrotum is governed by gubernacular regression. CSL regression is thought to be controlled by androgens produced by Leydig cells. Gubernacular outgrowth (and smooth-muscle-like cell differentiation, suggesting active contraction as the descent mechanism) is stimulated by Insl3 (insulin-like hormone 3), another product of Leydig cells. The nature of hypothetical Insl3 receptor in gubernaculum is not yet clear. It is thus interesting, that a receptor with unknown ligand, Great (G-protein coupled receptor affecting testis descent), is expressed in the gubernaculum and its mutation caused cryptorchidism both in mice and in humans. Another example of genetically determined cryptorchidism is targeted mutation in Hoxa10 (homeobox transcription faktor a10). However, this is not likely to be the cause of human idiopathic, non-syndromic cryptorchidism, because the mutation affects severely also female reproduction and, moreover, lumbal vertebrae show anterior homeotic transformation.
Testicular temperature is lower than the core body temperature. Prolonged exposure of the undescended testis to the increased temperature is for a long time believed to be the cause of compromised spermatogenesis in cryptorchidism. Early surgical reposition (orchiopexy) leads to normal testis development including fertility. On the other hand, examination of rodent testis exposed to a single heat stress or experimental cryptorchidism revealed apoptosis of pachytene spermatocytes as a mechanism of germ cell degeneration. Very similar pathology was found in testes of transgenic mice expressing human Hsf1 (heat shock transcription factor 1), a transcription regulator of heat shock proteins. One can hypothesize, that Hsf1 functions in testes as a temperature sensor, which drives spermatocytes, in contrast to other cell types, to death in case of elevated temperature to prevent genetically damaged germ cells to complete spermatogenesis. Pachytene stage is the period of crossing-over and DNA repair is necessary for error-free completion of recombination during crossing over. The cells have intrinsic quality checkpoint system, where cells with DNA damage die by apoptosis. Elevated temperature may harm by increasing number of dsDNA breaks.
Figures
Fig. 3.1. Sex determination in males and females
A: in males, Sry gene on Y-chromosome is expressed in somatic cells (brown) and upregulates Sox9. Sox9 then determines the fate of the somatic cells to become Sertoli cells. Sertoli cells 1) switch germ cells (yellow) to the spermatogonia program, 2) produce anti-Müllerian hormone, which destroys Müller duct and inhibits thus uterus and oviduct development and 3) induce differentiation of Leydig cells. Leydig cells have endocrine activity - they produce several hormones, some with highly specialized function, e. g. Insl3 (insulin-like 3), which through its probable receptor (Great) support development and contraction of gubernaculum, driving descent of testis. Main product of Leydig cells are steroid hormones, mainly testosterone, which acts through androgen receptor. Testosterone supports external genital development, persistence of Wolffian duct and drives pubertal development.
B: in females, germ cells differentiate into oogonia, which recruit follicular cells and proceed to meiosis. Feminine external genital develops and Wolffian duct disappears in absence of testosterone. In absence of AMH, Müller duct differentiates into uterus and oviducts.
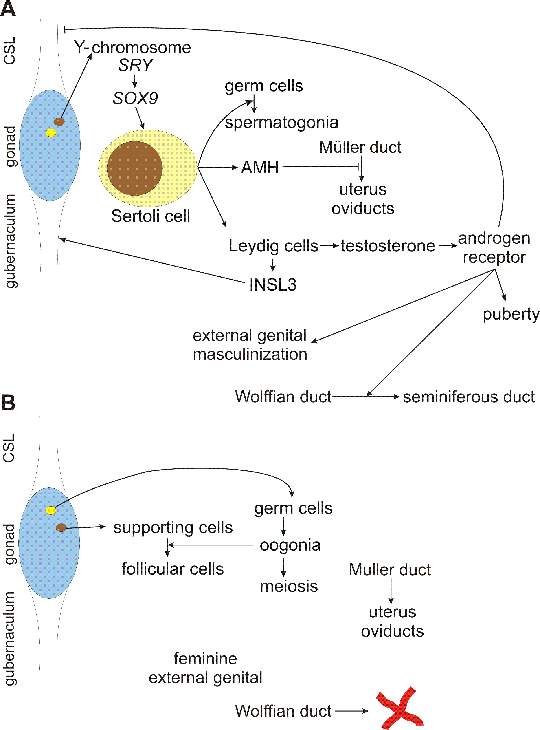
Fig. 3.2. Human Y chromosome
a: Y chromosome is the smallest human chromosome - length of typical human Y chromosome is estimated to about 57 Mb. However, only about half of the chromosome contains coding sequences, since Y chromosome contains the largest segment of constitutive heterochromatin, comprising almost entire q12 band. This heterochromatin segment segment is highly variable to such extent, that the different length of q12 band can be detected using classical cytogenetic methods, e. g. G-banding.
b: sex chromosomes evolved from ancient pair of homologous autosomes, with function and gene content similar to the current X-chromosome. Y chromosome evolution was initiated by two events - acquisition of male determining gene SRY and suppresion of recombination with X chromosome in the segment containing SRY (probably due to inversion or similar rearrangement). Suppression of recombination initiated sequence diversification, creating male-specific region, which consequently lost most of the genes. The genes are kept only on X chromosome - "Y chromosome as a decaying X chromosome". For damaged X chromosome can be repaired using the homologous chromosome, at least in female cells, but Y chromosome has no homologue in euploid male cells. However, pairing of X and Y chromosomes is essential in meiosis for proper segregation, so in addition to male specific region, Y chromosome retains two pseudoautosomal regions, PAR1 and PAR2 on both telomeric ends, which are identical with the appropriate telomeric segments of X chromosome. Therefore, the genes localized in PARs (about 14 genes in PAR1, about 3 genes in PAR2) exhibit autosomal pattern of inheritance. het. = heterochromatin. Heterochromatic regions of human chromosomes remained largely unsequenced. Y chromosome is rather an exception, because all euchromatin/heterochromatin boundaries were sequenced, as well as a portion of the centromere core. Heterochromatin is thought to consist of massively amplified degenerated tandem repeats of low sequence complexity. On Y, the centromeric region (cen) contain 171 bp alpha satellite type repeat, with 5941 bp secondary repeat unit and GGAAT type repeat. Yq12 contains GGAAT repeats and an AT rich repeat with about 3 kb unit.
c: male specific region of Y chromosome is a patchwork of several sequence classes - X-degenerate sequences are homologous to X chromosome, but the coding sequences are diverged and partially degraded. Hundreds of ancestral X-linked genes were completely deleted, 13 genes lost function by mutations and became pseudogenes. The remaining 16 genes are ubiquitously expressed; they are probably under selection pressure to remain active in two copies even in males. Polymorphism between X and Y copy of one of these genes, coding for enamel protein amelogenin is used for sex identification in the DNA fingerprinting set (CODIS = Combined DNA Index System). X-transposed sequences were relatively recently translocated from X chromosome, there are only 2 genes. Unique Y chromosome sequences comprise large inverted palindromes (pal.) and tandem repeated sequences (on Yp11.2), together also called "amplicons", with approximately 60 genes in multiple, nearly identical copies, that can be grouped into 9 families, each characterized by >98% nucleotide identity among family members, in both exons and introns. The genes are from diverse sources: VCY and RBMY families were, similar to the X-degenerate genes, derived from common ancestors of the X- and Y-chromosomes. Other genes, like DAZ were acquired through a series of autosomal transpositions. Yet another molecular mechanism accounts for the CDY genes, which arose by retrotransposition and subsequent amplification of a processed messenger RNA derived from an autosomal gene. Nearly all these genes are almost exclusively expressed in the spermatogenic cells of the testis. So the Y chromosome accumulated genes necessary for or improving spermatogenesis. Their amplification into families increases the probability of maintenance of their function in a region devoid of crossing over, by another type of homologous recombination, gene conversion among family members.
Mutation or deletion of SRY gene leads to male to female sex reversal. Deletion of other parts of Y chromosome can cause male infertility with azoospermia. There are 3 loci with recurrent deletions in azoospermic patients: AZFa, AZFb and AZFc. AZFc deletions are most frequent, azoospermia is probably caused by removal of all 4 DAZ (deleted in azoospermia) genes. DAZ gene product is an RNA-binding protein that is important for spermatogonia function.
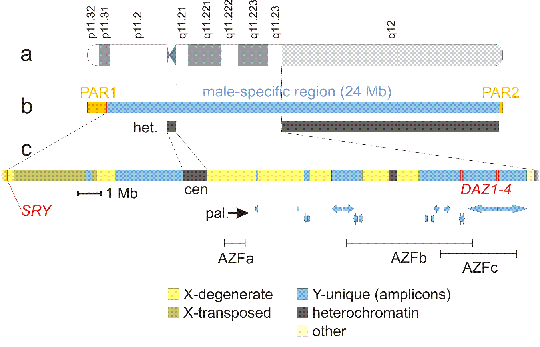
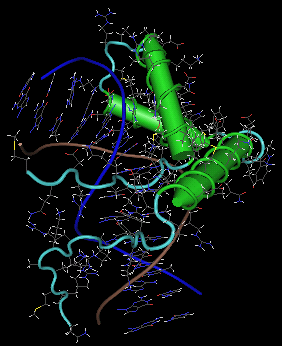
Fig. 3.3. Structure of SRY protein
Structure of SRY protein bound to a minor groove of specific DNA segment. DNA strands backbone is in blue and brown. Protein alpha helices are in green, loops in light blue. There is no beta sheet. Side chains of DNA as well as protein are colored as follows: C dark grey, O red, N blue, S yellow and H white. Note pronounced DNA bending - at the bottom the double helix has vertical orientation, but due to interaction with the protein, the helix bends to the left in the upper part. 3D structure can be retrieved from Protein Databank (PDB), from Entrez Structure (at NCBI) and from other databases under the accession 1J46 (direct link to the structure at PDB: http://www.pdb.org/pdb/explore/explore.do?structureId=1J46, or at NCBI: http://www.ncbi.nlm.nih.gov/Structure/mmdb/mmdbsrv.cgi?form=6&db=t&Dopt=s&uid=17220). The snapshot of the structure was created by Cn3D, freely available from NCBI. However, many other visualization tools exist and most are free - it is advisable to try several possibilities to get better insight into the structure