Aktuální genetika
Multimediální učebnice lékařské biologie, genetiky a genomikyNemendelovská dědičnost
Některá onemocnění a znaky, za které zodpovídají variace jednotlivých genů, nesledují do značné míry pravidla přenosu do dalších generací, která platí pro dědičnost "klasických" monogenních onemocnění, ať už autozomálně či gonozomálně determinovaných. Proto se tato různorodá skupina někdy vyděluje pod pojmem tzv. nemendelovské dědičnosti (angl. non-Mendelian inheritance). Řadíme sem především mitochondriální dědičnost, nestabilitu repetitivních sekvencí a genomický imprinting. Zcela zvláštní kapitolou je epigenetika (viz kapitola Epigenetika). Naopak se do této skupiny obvykle nepočítají jevy, které "komplikují" monogenní dědičnost, např. penetrance, expresivita znaku atd.
1. Mitochondrie a mitochondriální dědičnost
a) Mitochondriální DNA
Ačkoli je podstatná část lidského genomu uložena v jaderné DNA, 37 strukturních genů je kódováno v cirkulárním mitochondriálním genomu, který je tvořen 16569 páry bazí DNA (označované jako mtDNA). Třetina těchto genů kóduje podjednotky komplexů respiračního řetězce zodpovědného za produkci ATP v rámci tzv. oxidačně-fosforylačního (OXPHOS) systému (13 polypeptidů, zbylých 74 je kódováno jaderným genomem), zbylé pak zodpovídají za tvorbu specifických molekul RNA potřebných pro syntézu proteinů (22 transferových RNA a dvě ribosomální RNA). V cytoplazmě je přítomno několik set až tisíc kopií mtDNA, když v každé mitochondrii je přítomna řada molekul mtDNA. O homoplazmii mluvíme, když je přítomna jen jedna homogenní populace mtDNA (odpovídá stavu u drtivé většiny "normálních" jedinců). Frekvence mutací je u mitochondrií 10-20x vyšší než u jaderné DNA, což vede k heteroplazmii, tedy stavu, kdy je přítomna smíšená populace s více variantami mtDNA. Mnohé vlastnosti mitochondriální DNA podporují teorii o původu mitochondrií coby endosymbiontických bakterií proto-eukaryotických buněk před cca 1,5 miliardami let. Mezi nejmarkantnější z nich určitě patří cirkulární organizace genomu, absence histonů, absence intronů či diskrétní počátky replikace. Navíc se mitochondrie obratlovců i dalších organismů se vyznačují odchylkami od univerzálního genetického kódu - kodón UGA není terminační, ale kóduje aminokyselinu tryptofan, naopak kodóny AGA a AGG nekódují leucin, ale ukončují translaci a nakonec kodón AUA místo leucinu kóduje metionin. Biologicky jsou asi nejbližšími příbuznými Rickettsie (mj. původci skvrnitého tyfu), což jsou obligátní intracelulární parazité. Předpokládá se, že Rickettsie a mitochondrie mají společného předka, který prodělal přechod od autonomní existence k endosymbióze.
Mitochondriální DNA skutčně obsahuje jen jednu významnou "nekódující" sekvenci, takzvanou D-smyčku (z angl. D-loop), což je krátký úsek mtDNA, ve kterém je těžký řetězec vytěsňován fragmentem DNA (500-700 nukleotidů), komplementárním k řetězci lehkému (a v tomto úseku má tedy mitochondrie třířetězcovou DNA). Zde je počátek replikace tzv. těžkého řetězce (označovaného jako H z angl. heavy), zmiňovaný fragment funguje jako primer pro začátek replikace. Počátek replikace lehkého řetězce (L z angl. light) je umístěn mimo D-kličku, asi ve 2/3 mt DNA. Transkripty obou řetězců musí být rozštěpeny, aby se mohly uvolnit funkční RNA (rRNA, tRNA a mRNA).
Přehledné zpracování tematiky mitochondriální biologie (v češtině) je možné nalézt na stránkách http://mujweb.atlas.cz/Veda/mitochondrie/.
Patrně nejrozsáhlejší soubor informací o mitochondriálním genomu, mitochondriálních onemocněních a dalších souvisejících témat je možné nalézt na stránkách MITOMAP: A human mitochondrial genome database, http://www.mitomap.org/.
b) Mitochondriální dědičnost
Z hlediska genetiky je zásadní fakt, že mtDNA je předávána další generaci výhradně matkou (matroklinní dědičnost), když po oplodnění jsou zachovány pouze mitochondrie lidského vajíčka. To patrně není pouhým důsledkem nepoměru počtu mitochondrií lidského oocytu (cca 100 000) a spermie (50-70), ale předpokládá se aktivní proces, který po oplození zlikviduje mitochondrie paternálního původu. Tomu odpovídá i typický maternální přenos chorob způsobených mutacemi mtDNA v rodokmenu (Obr.1.). Pokud je heteroplazmická mutace zděděna nebo k ní dojde v časných fázích embryogeneze, normální i mutovaná varianta jsou náhodně předávány při buněčném dělení dceřinným buňkám (mitotická i meiotická segregace). Distribuce a zastoupení mutované mtDNA v jednotlivých orgánech jsou proto patrně závislé na čase a vzniku mutace a rovněž na typu postižené buňky.
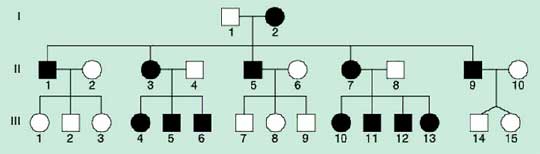
Obr.1. Typický rodokmen pro onemocnění s mitochondriální dědičností
Je třeba si uvědomit, že zdaleka ne veškerá onemocnění způsobená dysfunkcí mitochondrií mají mitochondriální typ dědičnosti. Pokud je defektní protein, jehož některé podjednotky kódují geny jaderného genomu a jiné zase přímo mtDNA, může být dědičnost zcela medndelistická. Sršeň a Sršňová uvádějí ve své monografii čtyři základní charakteristiky dědičných onemocnění, u kterých je třeba diferenciálně diagnosticky uvažovat o mitochondriální dědičnosti:
- Maternální typ dědičnosti (viz výše)
- Díky mitotické a meiotické segregaci nacházíme různý stupeň postižení v různých tkáních a variabilní projevy u potomků v jedné mateřské linii, což souvisí s existencí tzv. prahového efektu, což je pozorování, že pro manifestaci dysfunkce je nutné, aby byl překročen určitý poměr mitochondrií s mutantní a normální mtDNA (ve většině případů 60-90%).
- Projevy poruchy OXPHOS systému. Dědičná mitochondriální onemocnění většinou postihují tkáně s vysokými nároky na přísun energie, jako jsou neuromuskulární či centrální nervový systém. Mezi běžně pozorované symptomy chorob s mitochondriální dědičností patří encefalopatie, ataxie, spasticita, (kardio)myopatie, hluchota nebo diabetes mellitus.
- Rozdíl oproti gonozomálně dominantní dědičnosti vázané na chromozóm X je v absenci přenosu mitochondriálně vázaného onemocnění na potomstvo a signifikantní převahu postižených žen u X-vázané dominantní dědičnosti.
2. Nestabilita repetitivních sekvencí
Nestabilita repetitivních sekvencí (angl. repeat instability) je relativně nedávno objevený typ mutace, kterému je přičítána zodpovědnost za více než 40 především neurodegenerativních a neuromuskulárních onemocnění. Tyto mutace jsou někdy označovány jako dynamické, jelikož se jejich charakter (počet repetic) mění z generace na generaci a také v závislosti na typu tkáně. Naproti tomu "statické" mutace se dědí prakticky v nezměněné podobě. Nejčastější formou nestability repetitivních sekvencí jsou repetice trinukleotidů (viz kapitola Repetitivní DNA), popsána jsou i onemocnění asociovaná s opakováním tetranukleotidů (dystrophia myotonica 2), pentanukleotidů (spinocerebelární ataxie 10), ale i minisatelitních a megasatelitních sekvencí. U většiny chorob této skupiny platí, že čím větší počet repetic, tím závažnější je jejich průběh a časnější věk manifestace. Dalším charakteristickým rysem nestability repetitivních sekvencí je tzv. genetická (generační) anticipace, tj. možnost zvyšování počtu repetic při přenosu na další generaci, zpravidla je tento efekt specifický u jednotlivých chorob pro potomky rodičů určitého pohlaví. Tímto mechanismem může dojít k tomu, že u dětí zdravých osob s "podprahově" zvýšeným počtem repetic (nositelů tzv. premutace) dojde v důsledku zmnožení repetic k manifestaci choroby.
Prvním popsaným onemocněním této skupiny je syndrom fragilního (chromozómu) X, který je obecně druhou nejčastější příčinou vrozené retardace mentálního vývoje (hned za Downovým syndromem). Fenotyp tohoto onemocnění je velmi variabilní, kromě mentálního deficitu různého stupně mohou postižení jedinci vykazovat tyto znaky: poruchy chování připomínající autismus, vysoký hlas, prolaps mitrální chlopně, makrocefalii, charakteristicky protažený obličej s prominující dolní čelistí, velké ušní boltce, u postižených mužů makroorchidismus (zvětšení varlat). Ač byl tento syndrom poprvé popsán v roce 1943 (Martin et Bell), své jméno získal díky pozorování Herberta Lubse, který v roce 1969 pozoroval sekundární konstrikce dlouhého raménka chromozómu X u několika mentálně retardovaných mužů, tzv. fragilní místo (Xq27.3). Molekulární podstatou většiny případů syndromu fragilního chromozomu X je expanze trinukleotidu CGG v 5'-UTR genu FMR1 (Fragile X Mental Retardation 1), která vede k útlumu transkripce a následnému chybění produktu FMR1 genu, proteinu FMRP. O tzv. plné mutaci (full mutation) mluvíme, pokud počet repetic převýší 200, normální počet repetic je 6-50. Jako premutace se označuje počet opakování mezi 60-200, nedávno se ovšem zjistilo, že tato varianta je zodpovědná za jiný syndrom, nazvaný fragile X tremor/ataxia syndrome (FXTAS). Nepřítomnost proteinu FMRP souvisí s celo řadou patofyziologických pochodů především v nervovém systému, jako příklad alespoň uveďme přítomnost četných dlouhých "nezralých" dendritů a defektní přenos signálu na postsynaptické úrovni u metabotropních glutamátových receptorů.
Friedreichova ataxie (FA) je nejčastější dědičná forma ataxie. Většina pacientů má homozygotní expanzi (GAA)n v prvním intronu genu pro frataxin, v důsledku čehož dochází ke snížení množství odpovídající mRNA a proteinu. Čím více trinukleotidových repetic je v genu přítomno, tím méně mRNA a proteinu je produkováno. Snížení koncentrací frataxinu je provázeno akumulací železa v mitochondriích, což vede ke zvýšené citlivosti na oxidační stres a snížení oxidativní fosforylace.
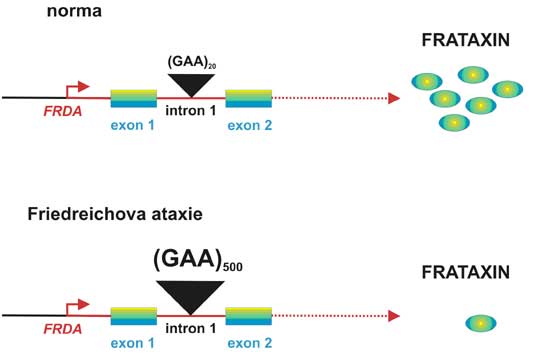
Obr.2. Upraveno podle Gatchel a Zhogbi, 2005.
3. Genomický imprinting
Genomický imprinting je proces, kdy je aktivita určitého genu regulována v závislosti na tom, od kterého rodiče byl gen zděděn. Ve většině případů to znamená, že pouze maternální nebo paternální alela je exprimována (je aktivní), kdežto druhá je utlumena. Mnoho našich současných znalostí o imprintingu pochází ze studií na myších. U těch, které sice měly kompletní sadu chromozómů, ale jeden pár chromozómů pocházel od jediného rodiče (uniparentální disomie), byly často popisovány abnormality vývoje a výskyt různých onemocnění. Zajímavé bylo, že to platilo jen pro některé chromozómy, v řadě případů se uniparentální disomie obešla bez zjevných následků pro svého nositele. V současnosti je u člověka známo zhruba osmdesát. imprintovaných genů a jejich počet roste. Většinou se v genomu nenacházejí tyto geny izolovaně, ale naopak se shlukují a je tak možné identifikovat celé chromozomální oblasti s imprintovanými geny. Zde nacházíme zásadní strukturu pro samotnou realizaci imprintingu, tzv. oblast řídící imprinting (imprinting control region - ICR). ICR jsou přístupné epigenetickým modifikacím a obvykle jedna z rodičovských ICR je metylována. Onemocnění, která vznikají na základě poruchy imprintingu, mají řadu příčin na molekulární úrovni, např. (mikro)delece, bodové mutace či uniparentální disomie. Mezi choroby s prokázanou úlohou poruch imprintingu patří syndromy Prader-Willi (OMIM 176270), Angelman (OMIM 105830), Beckwith-Wiedemann (OMIM 130650), Silver-Russell (OMIM 180860), transientní novorozenecký diabetes mellitus (OMIM 601410) a další.
Zdroje a další čtení - česky:
- Thompson and Thompson, Klinická genetika, TRITON 2004, str.232-238.
- Seemanová E. SYNDROMY A ONEMOCNĚNÍ Z MUTACÍ TYPU ZMNOŽENÍ TRINUKLEOTIDŮ Čas. Lék. čes., 141, 2002, No. 16, p. 503-507.
Zdroje a další čtení - ostatní:
- Sršeň, Š., Sršňová, K. Základy klinickej genetiky a jej molekulárna podstata. Osveta 200 Nature Rev Genet. 2005
- Genetics of developmental disabilities. Butler MG., Meaney F.J. eds.. Taylor & Francis Group, LLC. 2005
- MITOMAP: A Human Mitochondrial Genome Database. http://www.mitomap.org, 2005.