Aktuální genetika
Multimediální učebnice lékařské biologie, genetiky a genomikyÚvod do proteomiky
Proteomika je obor, který se zabývá globálním hodnocením exprese genetické informace na úrovni bílkovin (proteomem), rovněž však zkoumá strukturu a interakce proteinů.
Human Proteome Organization (HUPO, www.hupo.org) formulovala v roce 2001 cíle proteomiky poněkud přesněji jako identifikaci všech proteinů kodovaných lidským genomem (popřípadě genomy dalších, zejména tzv. modelových organismů) s následným stanovením a) jejich exprese v různých buňkách daného organismu - expresní proteomika, b) jejich subcelulární lokalizace v různých organelách, c) jejich posttranslačních modifikací, d) jejich vzájemných interakcí (b-d zahrnuje strukturní proteomika) a e) vztahu mezi strukturou a funkcí (funkční proteomika).

Z této širokého záběru obou definic definice je jasné, že proteomika má řadu dílčích podoborů, např. tzv. klinická proteomika se pomocí analýzy bílkovinného spektra vzorků (např. moč, sliny, krev) pokouší odhalit nové markery, které by v budoucnosti mohly pomoci ve včasné diagnóze a léčbě konkrétního onemocnění. Naprosto zásadní roli ve všech sférách proteomiky hraje bioinformatika, ať už ve formě digitalizace a obrazové analýzy 2D gelů (viz níže), modelování trojrozměrných struktur proteinů a sítí interakcí proteinů nebo ve formě tvorby, správy a vývoje veřejně přístupných databází. Jedním ze zajímavých projektů je např. švédský Human Protein Atlas (http://www.proteinatlas.org/) , který shromažďuje informace o expresi jednotlivých proteinů v různých tkáních, což by samo o sobě nebylo tak unikátní, ale zároveň pro tyto proteiny archivuje snímky imunohistochemicky barvených preparátů - v době psaní tohoto textu jich bylo k dispozici přes 400 000.
I když by se mohlo zdát, že za pomoci dnes již poměrně dostupných RNA/cDNA "čipů" (microarray) hodnotících expresi na úrovni transkriptů získáváme dostatečně detailní informaci o aktuálně probíhajícím stavu realizace genetické informace, je třeba si uvědomit, že:
- ne všechny mRNA jsou určeny pro překlad do proteinu
- míra transkripce konkrétní, protein-kódující RNA zdaleka ne vždy odpovídá míře exprese či dokonce aktivity kódovaného proteinu díky mnoha faktorům (mRNA, RNA sestřih, posttranslační modifikace proteinů, atd.)
Stejně jako u ostatních "-omik", i rozvoj proteomiky byl zásadním způsobem ovlivněn vývojem na poli technologie. Reálný rozvoj oboru nastal de facto teprve po té, co k dnes již klasické metodě dvojrozměrné elektroforézy se přidal rozvoj "měkkých" ionizačních technik hmotnostní spektrometrie, kam se řadí desorpce/ionizace za účasti matrice (MALDI) a ionizace elektrosprejem (ESI).
Obrázek 1: Hlavní směry proteomiky
Metody proteomiky
1. Dvojrozměrná elektroforéza proteinů
V současné době je dvojrozměnrná elektroforéza proteinů metdou volby pro separaci proteinů. Tato metoda umožňuje rozlišení až 10000 proteinů. Podstatou metody je využití dvou odlišných fyzikálně chemických vlastností proteinů: nejdříve jsou proteiny rozděleny podle jejich izoelektrického bodu (pI), což je hodnta pH, při které je součet nábojů proteinu nulový. Toho docílíme tak, že na proužek gelu, ve kterém je vytvořent gradient pH, naneseme protein a aplikujeme elektrické napětí. Proteiny pak migrují ke katodě či anodě dle svého celkového náboje až do chvíle, kdy pH místa v gelu odpovídá pI proteinu. Podle zvoleného rozsahu pH můžeme měnit škálu proteinů, které rozdělíme, často se právě této vlastnosti využívá pro "zaostření" (zooming) na proteiny s pI v úzkém rozsahu pH. Po separaci v prvním rozměru je provedena běžná elektroforéza na polyakrylamidovém gelu (PAGE), kdy ovšem napětí aplikujeme kolmo vzhledem k původní orientaci elektrod. Proteiny poté migrují v druhém rozměru glelem čistě v závislosti na jejich velikosti. Po proběhnutí obou fází 2D elektroforézy je třeba proteiny vizualizovat některou z barvících či značících metod (chemických nebo radioaktivních). Výsledné "mapy" proteinů lze porovnávát např. mezi experimentálním a kontrolním vzorkem nebo mezi vzorky odebreanými od pacientů s konkrétním onemocněním oproti zdravým kontrolám a identifikovat tak odlišně exprimované proteiny, které mohou mít souvislost s patogenezí daného onemocnění. Proto je třeba ověřit identitu těchto odlišně exprimovaných proteinů, nejčastěji pomocí "vyříznutí" oblasti gelu vykazující odlišnost a její následnou analýzu pomocí hmotnostní spektrometrie.
Metoda 2D elektroforézy má samozřejmě i své nevýhody - patří mezi ně omezená reprezentativnost vzorku (problematické jsou velmi zásadité proteiny, proteiny málo rozpustné ve vodné fázi a membránové proteiny), sensitivita (proteiny přítomné ve velmi nízkých koncentracích vyžadují speciální typy barvení), reproducibilita a ve srovnání s obdobnými technikami pro nukleové kyseliny špatná automatizovatelnost.
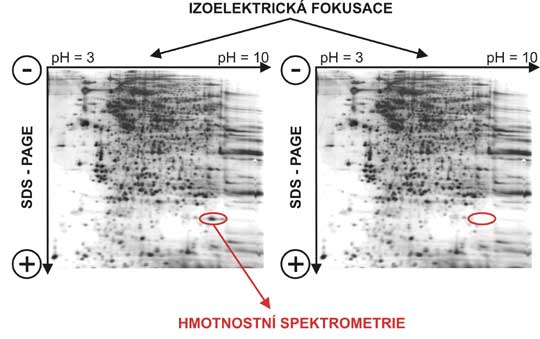
Obrázek 2. Dvojrozměrná (2D) elektroforéza. Po izoelektrické fokusaci v pH gradientu v elektrickém poli následuje elektroforetická separace v polyakrylamidovém gelu (SDS-PAGE...sodium dodecylsulphate - polyacrylamide gele electrophoresis). Vizualizace proteinů je možná po obarvení (na obrázku použita Coomasie blue).
2. Hmotnostní spektrometrie
Hmotnostní spektrometrie je metoda, která umožňuje přesné měření molekulární hmotnosti široké škály látek. Jelikož zkoumaná látka musí být převedena jako intaktní do plynné fáze, využití hmotnostní spektrometrie pro analýzu proteinů (ale i polysacharidů či oligobukleotidů) bylo umožněno vývojem "měkkých" ionizačních technik hmotnostní spektrometrie, kam se řadí desorpce/ionizace za účasti matrice (MALDI) a ionizace elektrosprejem (ESI). Identifikace proteinu se provádí v zásadě dvěma základními způsoby:
- Protein je naštěpen trypsinem nebo jiným proteolytickým enzymem ne menší peptidy, jejichž přesné hmotnosti jsou pomocí MS změřeny. Spektrum těchto hmotností je pak porovnáno s teoretickými spektry, která jsou vypočítána ze sekvencí proteinů v dsostupných databázích (pomocí bioinformatických metod).
- Tandemová MS umožňuje zvolit peptid, který je následně fragmentován kolizí s inertním plynem. Profil výsledků fragmentace (fragmentation pattern) poskytuje částečnou či úplnou informaci o sekvenci proteinu, která je vodítkem pro hledání shody s daty uloženými v databázích.
Mimo samotnou identifikaci proteinů je MS neocenitelným nástrojem pro analýzu postranslačních modifikací proteinů, protože umožňuje lokalizaci dané modifikace uvnitř proteinu a napomáhá rovněž zjištění charakteru takové modifikace.
Proteinový čip
Ve srovnání s nukleovými kyselinami je díky daleko větší variabilitě fyzikálně-chemických vlastností proteinů konstrukce jednotného "proteinového čipu" (protein array) daleko komplikovanější. I přesto je v současnosti k dispozici několik platforem, které se k tomuto cílí úspěšně blíží. Některé využívají protilátky nebo antigeny, které jsou na čipu naneseny ve vysoké denzitě a ve vzorku detekují odpovídající entitu, jiné pro změnu obsahují neproteinové polymerní molekuly, které reagují s určitými skupinami proteinů, hodně naděje je vkládáno do nastupujících nanotechnologických metod.
Proteomika v medicíně
Jak bylo naznačeno výše, jednou z podstatných úloh proteomiky je identifikace nových markerů využitelných pro predikci, prevenci, diagnózu, prognózu a optimalizaci terapie lidských onemocnění. Aby takové využití metody bylo skutečně reálné v každodenní klinické praxi, je nutné, aby biologický materiál, ve kterém bude takový marker identifikován, byl poměrně snadno dostupný (např. z krve, moče, slin, mozkomíšního moku apod.). Další, prozatím spíše experimentální využití, bude pravděpodobně v oblastech vývoje léčiv a farmakoproteomiky.
Zdroje / Další literatura
- ÚHKT - Materiály k přednáškám proteomika 2004
http://www.uhkt.cz/vyuka/proteomika - Proteomická sekce ČSBMB Co je proteomika?
http://proteom.biomed.cas.cz/proteomics/proteomics.cs.php - Molecular Biologist's Guide to Proteomics
Molecular Biologist's Guide to Proteomics